Het myelodysplastisch syndroom (MDS) betreft een heterogene groep ziektebeelden die zich kenmerken door een inefficiënte hematopoëse resulterend in één of meerdere cytopenieën waarbij bij een deel van de patiënten progressie optreedt naar Acute Myeloide Leukemie (AML). MDS kan de novo ontstaan of secundair na voorafgaande chemo- en/of radiotherapie.. Afhankelijk van het blasten percentage, de cytopenie en de aanwezigheid van cytogenetische afwijkingen is een prognostische classificatie mogelijk. Afhankelijk hiervan kan de therapie variëren van een afwachtend beleid tot intensieve behandeling al dan niet in combinatie met allogene stamceltransplantatie.
Diagnostiek van MDS (altijd gebaseerd op bloed en beenmerg (BM) onderzoek)
Een in de klinische praktijk bruikbare definitie van MDS wordt gevormd door de volgende twee voorwaardelijke criteria:
- Aanhoudende cytopenie in 1 of meer cellijnen gedurende ten minste 6 maanden (Hb < 6.8, neutrofielen < 1,5 x 109/L, trombocyten < 100 x 109/L)
- Uitsluiting van andere (niet) klonale hematologische of andere niet-hematologische aandoening.
Daarnaast tenminste 1 van 3 additionele criteria:
- Morfologische dysplasie in ≥ 10% erytroide cellen, neutrofielen en voorlopers, en/of megakaryocyten. Of : > 15% ringsideroblasten
- BM blasten 5-19%
- Voor MDS specifieke cytogenetische afwijking (zie tabel)
| Unbalanced abnormalities | Balanced abnormalities |
| −7 or del(7q) | t(11;16)(q23;p13.3) |
| −5 or del(5q) | t(3;21)(q26.2;q22.1) |
| i(17q) or t(17p) | t(1;3)(p36.3;q21.1) |
| −13 or del(13q) | t(2;11)(p21;q23) |
| del(11q) | inv(3)(q21q26.2) |
| del(12p) or t(12p) | t(6;9)(p23;q34) |
| del(9q) | |
| idic(X)(q13) | |
| Complex karyotype (3 or more chromosomal abnormalities) involving one or more of the above abnormalities | |
Moleculaire afwijkingen (mutaties) kunnen richtinggevend zijn voor de diagnose MDS maar dienen met terughoudendheid geinterpreteerd te worden omdat ze ook voorkomen in het kader van veroudering (‘CHIP’) of aplastische anemie
Literatuur:
Valent P, Horny HP, Bennett JM, et al. Definitions and standards in the diagnosis and treatment of the myelodysplastic syndromes: consensus statements and report from a working conference. Leuk Res 2007;31:727–36
Platzbecker U, Sanini V,Mufti GJ, et al Update on developments in the diagnosis and prognostic evaluation of patients with myelodysplastic syndromes (MDS) consensus statements and report from an expert workshop. Leuk.Res.2012, 36(3)264-70
Onderzoek bij diagnose
- Bloed: Hb, Ht, erytrocyten, celindices, reticulocyten, leukocyten, leukocyten differentiatie, trombocyten en morfologie perifeer bloed
- Kreatinine, bilirubine, AF, γGT, ALAT, ASAT, LD, Fe, ferritine, transferrine, totaal eiwit, albumine, evt M proteine
- Coombs, haptoglobine, vitamine B12, foliumzuur,
- Virus serologie: Parvo B19, op indicatie HIV, CMV, Hepatitis B/C
- PNH
- HLA-DR15* op indicatie (*hypoplastische MDS)
- serum koper.
- Bij bewezen MDS: Erytropoietine (EPO) spiegel
- Beenmerg: morfologie, immunologie, cytogenetica, cryopreservati biobank bij informed consent (NB moleculair onderzoek is nog geen standaard tenzij CMML, maar wordt sterk aanbevolen)
- (CMML/MDS/MPN: Echo-bovenbuik (lever, milt in cm).
- Botbiopt
Moleculaire diagnostiek
Moleculaire diagnostiek kan zowel diagnostische als prognostische betekenis hebben in MDS.
Diagnostische waarde: Als er een SF3B1 mutatie wordt aangetoond dan is een percentage van 5% ringsideroblasten voldoende om de diagnose MDS-RS te stellen (in plaats van 15% ringsideroblasten)
Prognostische waarde: Mutaties aangetoond met NGS kunnen in aanvulling op de IPSS-R prognostische betekenis hebben, waarbij met name mutaties in TP53 en het aantal mutaties (>3) geassocieerd zijn met een slechtere prognose en kortere duur van respons bij hypomethylerende therapie. Ongunstig zijn verder mutaties in ASXL1, ETV6, EZH2, en RUNX1. Relatief gunstig zijn mutaties in SF3B1.
Classificaties van myelodysplastische syndromen
De volgende morfologische classificaties worden gebruikt:
WHO-classsificatie 2016
| Name | Dysplastic lineages |
Cytopenias * | Ring sideroblasts as % of marrow erythroid elements | BM and PB blasts | Cytogenetics by conventional karyotype analysis |
| MDS with single lineage dysplasia (MDS-SLD) |
1 | 1 or 2 | <15%/<5% ¶ | BM <5%, PB <1% no Auer rods |
Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS with multilineage dysplasia (MDS-MLD) |
2 or 3 | 1-3 | <15%/<5% ¶ | BM <5%, PB <1% no Auer rods |
Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS with ringsideroblasts (MDS-RS) |
|||||
| MDS-RS with single lineage dysplasia (MDS-RS-SLD) |
1 | 1 or 2 | ≥15%/≥5% ¶ | BM <5%, PB <1% no Auer rods |
Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS-RS with mulitlineage dysplasia (MDS-RS-MLD) |
2 or 3 | 1-3 | ≥15%/≥5% ¶ | BM <5%, PB <1% no Auer rods |
Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS with isolated del(5q) | 1-3 | 1-2 | None or any | BM <5%, PB <1% no Auer rods |
del(5q) alone or with 1 additional abnormality except -7 or del(7q) |
| MDS with excess blasts (MDS-EB) |
|||||
| MDS-EB1 | 0-3 | 1-3 | None or any | BM 5%-9% or PB 2%-4%, no Auer rods | Any |
| MDS-EB2 | 0-3 | 1-3 | None or any | BM 10-19% or PB 5%-19% or Auer rods | Any |
| MDS unclassifiable (MDS-U) | |||||
| with 1% blasts | 1-3 | 1-3 | None or any | BM <5%, PB = 1% ∑, no Auer rods | Any |
| with single lineage dysplasia and pancytopenia | 1 | 3 | None or any | BM <5%, PB <1%, no Auer rods | Any |
| based on defining cytogenetic abnormality | 0 | 1-3 | <15% § | BM <5%, PB <1%, no Auer rods | MDS-defining abnormality |
| Refractory cytopenia of childhood | 1-3 | 1-3 | None | BM <5%, PB <2% | Any |
| * Cytopenias defined as: hemoglobin<10 g/dL; platelet count<100 x 109/L; and absolute neutrophil count<1.8 x 109/L. Rarely, MDS may present with mild anemia or thrombocytopenia above these levels. PB monocytes must be <1 x 109/L ¶ If SF3B1 mutation is present. Σ One percent PB blasts must be recorded on at least 2 separate occasions. § Cases with ≥15% ring sideroblasts by definition have significant erythroid dysplasia, and are calssified as MDS-RS-SLD. |
|||||
Speciale vormen van MDS:
- Hypoplastische MDS
- MDS met fibrose (MDS-F)
- Therapie-gerelateerde MDS (t-MDS)
MDS Risicoclassificatie
Behandeling
Zeer laag/laag risico MDS (volgens IPSS-R)
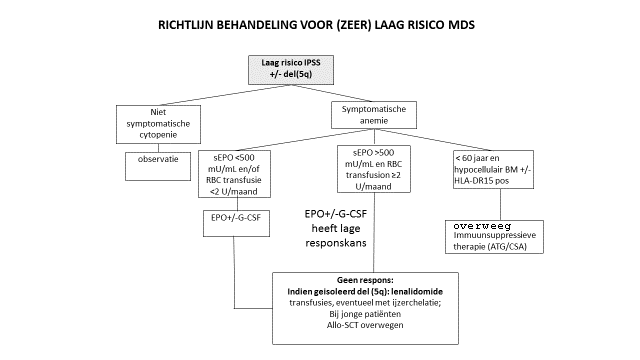
*) NB: Indien slecht risico cytogenetica en/of blasten tot 10%: bespreek Allo-SCT.
Behandeling met EPO/G-CSF
Een behandeling met Erytropoietine (Epo) en G-CSF kan worden overwogen afhankelijk van de Epo spiegel en transfusie behoefte. De kans op eventuele respons en mate van respons kan bepaald worden volgens onderstaande criteria:

Indien besloten wordt tot behandeling met EPO/G-CSF kan onderstaand schema worden gebruikt.

NB: Bij falen: nagaan Fe, Foliumzuur, BM (status ziekte).
Lenalidomide is goedgekeurd voor de behandeling van patiënten met transfusie-afhankelijke anemie als gevolg van laag of intermediair-1 risico (IPSS) myelodysplastische syndromen (MDS) geassocieerd met een geïsoleerde 5q-deletie wanneer andere therapeutische opties onvoldoende of inadequaat zijn (1dd 10 mg dag 1-21 herhalen dag 28).
Studies voor patiënten met zeer laag/laag/intermediair risico MDS
ACE-536-MDS-002: Celgene Ace-536-MDS-002 (COMMANDS trial)/A Phase 3, Open-Label, Randomized Study to Compare the Efficacy and Safety of Luspatercept (ACE-536) versus Epoetin Alfa for the Treatment of Anemia due to IPSS-R Very Low, Low or Intermediate Risk Myelodysplastic Syndromes.
Inclusiecriteria:
- Zeer laag, laag of intermediair risico MDS (IPSS-R); EPO <500 U/L
- Erythrocyten transfusie afhankelijk (2-6E per 8 weken)
- voorafgaand maximaal 2 giften EPO (geen darbepoëtine), laatste dosis ≥ 8 weken geleden
Intermediair risico (volgens IPSS-R)
Intermediair MDS (R-IPSS 3-4,5) kan behandeld worden volgens zeer laag en laag risico MDS of volgens hoog en zeer hoog risico MDS, afhankelijk van ziektekenmerken en patiënt factoren (waaronder leeftijd en HCT-CI score).
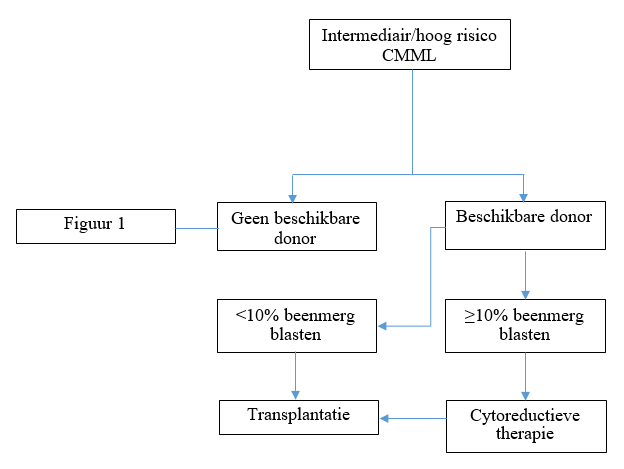
NB: Bij slecht risico cytogenetica en/of toename blasten > 10%: behandeling volgens hoog-risico algoritme.
Hoog of zeer hoog risico (volgens IPSS-R)
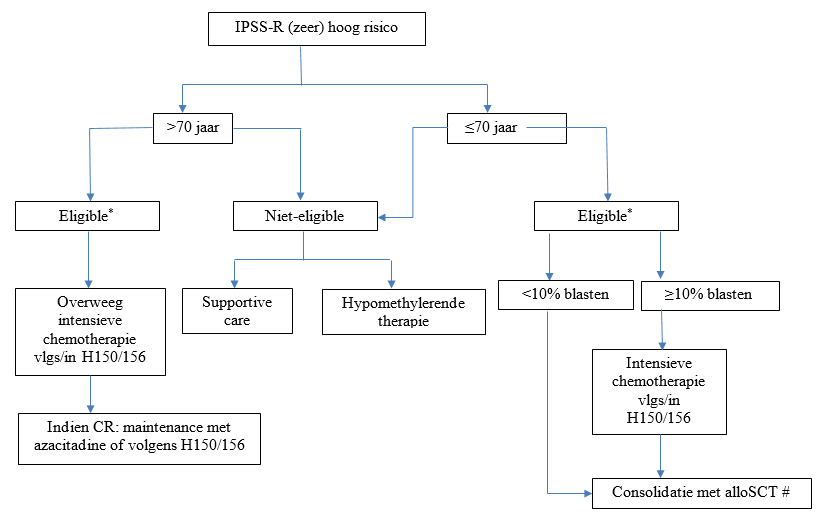
* Indien slecht risico genetica (bijv. TP53 mutatie, monosomaal karyotype) overweeg hypomethylerende therapie i.p.v. intensieve chemotherapie
# Bij MDS-EB2 patiënten die intensieve remissie-inductie ondergaan, dient bepaling van de optimale consolidatie (allo-SCT versus auto-SCT) volgens het ELN algoritme (zie vademecum AML: consolidatie) te geschieden.
NB: MDS < 10% blasten (BM), intermediair/hoog of zeer hoog risico volgens IPSS-R, kunnen in de eerste lijn direct allogeen getransplanteerd worden (zie transplantatie indicaties)
Azacytidine (Vidaza) is geregistreerd voor de volgende indicaties:
intermediair 2 en hoog risico myelodysplastische syndromen (MDS) volgens het International Prognostic Scoring System (IPSS), chronische myelomonocytaire leukemie (CMML) met 10-29% beenmergblasten zonder myeloproliferatieve aandoening, acute myeloïde leukemie (AML) met 20-30% blasten en multilineaire dysplasie, volgens de indeling van de Wereldgezondheidsorganisatie (WHO) en AML met >30% beenmergblasten volgens de indeling van de WHO.Dosis 75 mg/m2 subcutaan dag 1 t/m 7. De kuur herhalen op dag 28. NB respons treedt in het algemeen pas op na 4-6 kuren.
Studies voor patiënten met hoog/zeer hoog risico MDS
Patiënten met een MDS-EB2 (WHO 2016) en een FLT3 of IDH1/2 mutatie kunnen, indien eligible, behandeld worden in de HOVON156 / HOVON150. zie KMS
Algemeen
Bij patiënten met ferritine > 1000 en regelmatig transfusies en levensverwachting > 3 jaar ijzerchelatie geven: (Zie hoofdstuk IJzerstapeling)
Bij uitgesproken granulopenie (neutro < 0,2) en recidiverende infecties antibiotische profylaxe overwegen. Schimmelprofylaxe indien neutro < 0,2.
CMML
Chronic myelomonocytic leukemia: CMML wordt in de WHO 2016 gerangschikt onder Myelodysplastische/myeloproliferatieve aandoeningen en heeft zowel dysplastische als myeloproliferatieve kenmerken.
De ziekte kenmerkt zich door:
- een persisterende monocytose (≥1x 109/l) in het perifere bloed >3 maanden
- het niet voldoen aan criteria voor BCR-ABL positieve CML, primaire myelofibrose, polycythemia vera en essentiële trombocytose
- afwezigheid van PDGFRA; PDGFRB of FGFR1 en PCM1-JAK2 gen herschikkingen
- <20% blasten in het perifere bloed en beenmerg
- dysplasie in tenminste 1 cellijn
óf
indien geen/minimale myelodysplasie en er is voldaan aan criteria 1 t/m 4 én:
-
- een verworven, klonaal cytogenetisch of moleculair-genetische afwijking in de hematopoietische cellen (bv TET2, SRSF2, ASXL1 en/of SETBP1)
- persisterende monocytose ≥3 maanden en alle andere oorzaken voor monocytose zijn uitgesloten (maligniteit, infectie en inflammatie)
Onderscheid wordt gemaakt tussen:
- CMML-0: in het bloed <2% blasten; in het beenmerg <5% blasten
- CMML-1: in het bloed 2-4 % blasten (inclusief promonocyten) en/of in het beenmerg 5-9% blasten
- CMML-2: in het bloed 5-19% blasten of 10-19% blasten in het beenmerg en/of aanwezigheid van Auerse staven
Diagnostiek: (als bij MDS)
- Hb, Ht, erytrocyten, celindices, reticulocyten, leukocyten, leukocyten differentiatie, trombocyten en morfologie perifeer bloed
- kreatinine, bilirubine, AF, γGT, ALAT, ASAT, LD, Fe, ferritine, transferrine, totaal eiwit, albumine, evt. M-proteine, ANF
- Coombs, haptoglobine, vit. B12, foliumzuur,
- bij bewezen MDS : Erytropoietine (EPO) spiegel
- virus serologie: Parvo B19, op indicatie HIV, CMV, Hep B/C, EBV, VZV, Arbo, Brucella, Leishmania, TBC
- Cytogenetica BM (inclusief FISH BCR-ABL)
- Moleculaire diagnostiek: bloed en BM (inclusief ASXL1 mutatie)
- bij eosinofilie ook PDGFRA/PDGFRB re-arrangements (via cytogenetica)
Risicoclassificatie CMML
Bij CMML-1 en CMML-2 wordt het risico vastgesteld op basis van de *Itzykson score:
| Leeftijd > 65 | 2 punten |
| WBC > 15x 109/l | 3 punten |
| HB < 6.2 mmol/l vrouwen | 2 punten |
| Hb < 6.8 mmol/l mannen | 2 punten |
| Trombocyten <100 x 109/l | 2 punten |
| ASXL-1 mutatie | 2 punten |
| Risico | Overall Survival (mediaan) * | AML-free survival (mediaan) * | |
|---|---|---|---|
| *Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol 2013; 31: 2428–2436. | |||
| Laag risico | 0-4 punten | 56 mnd | |
| Intermediair risico | 5-7 punten | 38.5 mnd | 27.4 mnd |
| Hoog risico | 8-12 punten | 14.4 mnd | 9.2 mnd |
Behandeling CMML
In 2018 zijn Europese aanbevelingen verschenen voor de behandeling van CMML (Itzykson et al, Hemasphere 2018). Deze worden hier in grote lijnen gevolgd.
Voor patiënten met CMML is de behandeling afhankelijk van de symptomatologie, de risicoscore en de leeftijd.
CMML patiënten die geen of slechts milde symptomatologie (milde cytopenie zonder myeloproliferatieve kenmerken) vertonen, hoeven niet noodzakelijkerwijze behandeld te worden.
Na bepalen van de risicoclassificatie is het van belang de eligibility van de patiënt voor allogene stamceltransplantatie vast te stellen.
Figuur 1: eligibility voor allogene stamceltransplantatie
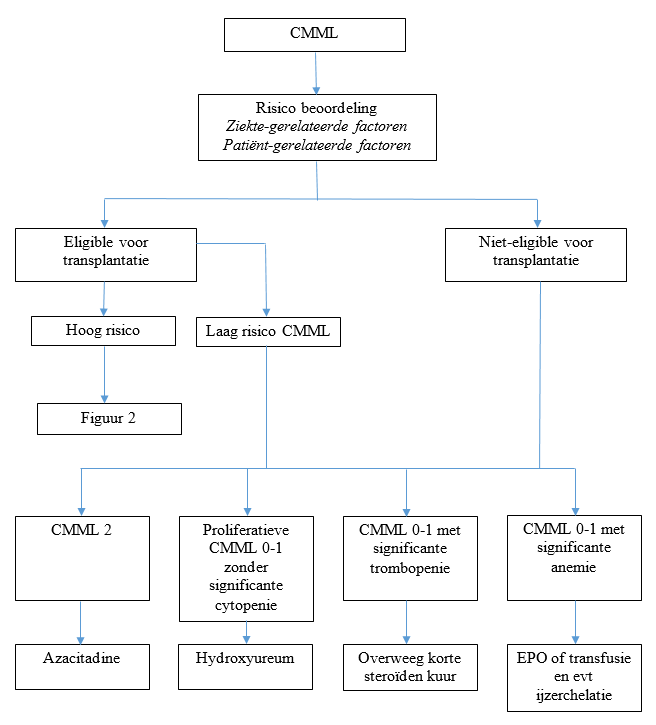
- Azacitidine 75 mg/m2 sc dag 1-7 elke 28 dagen.
- Hydrea bij forse splenomegalie
Laag risico CMML patiënten:
CMML patiënten met een laag risico komen in principe niet in aanmerking voor behandeling met een allogene SCT.
Indien slechte risico kenmerken* een rol spelen, kan behandeling dan wel allogene stamceltransplantatie overwogen worden.
* slechte risico kenmerken gedefinieerd als:
slecht risico genetica, toename of persisterend verhoogd aantal blasten (>50% of > 15% beenmerg blasten), levensbedreigende cytopenieën, hoge transfusie behoefte, slecht-risico moleculaire kenmerken
Intermediair en hoog risico CMML patienten:
- overweeg allogene stamcel transplantatie, wel of niet voorafgegaan door inductie chemotherapie volgens Hovon 150/156
Figuur 2: therapeutisch algoritme voor intermediair en hoog-risico CMML patiënten